
|
|
|
for Energy, Catalysis and Biomolecular Applications |
|---|
The research activity is centered on first-principles simulations of surfaces (in the form of model planar systems or of the surface of nanomaterial), of the interaction and organization of molecules at these surfaces and of their chemistry and catalytic reactivity. Applications are focused on the sustainable transformation of energy and resources. A large part of the activity aims at understanding molecular reactivity on the surface of heterogeneous catalysts from a computational chemistry approach.
Our group pioneered the structure-based activity descriptors for heterogeneous catalysts (see publications below). The obtained fully predictable, structure-sensitive scaling relations are a major step towards the fast screening of metallic catalysts for a given reaction. The method is particularly powerful for describing the catalytic activity on nanoparticles. The proposed a new descriptor called "generalized coordination" scales linearly with adsorption energy for various site structures and also for different particle sizes and enables the design of the optimal structure for a catalyst active site. It was predicted by the model and confirmed by experiments that small cavities at the Pt(111) surface enable to increase the activity for oxygen reduction reaction by a factor 3.5. Catalyst optimization through particle size and condition-dependent morphology is the key target of this branch of research.
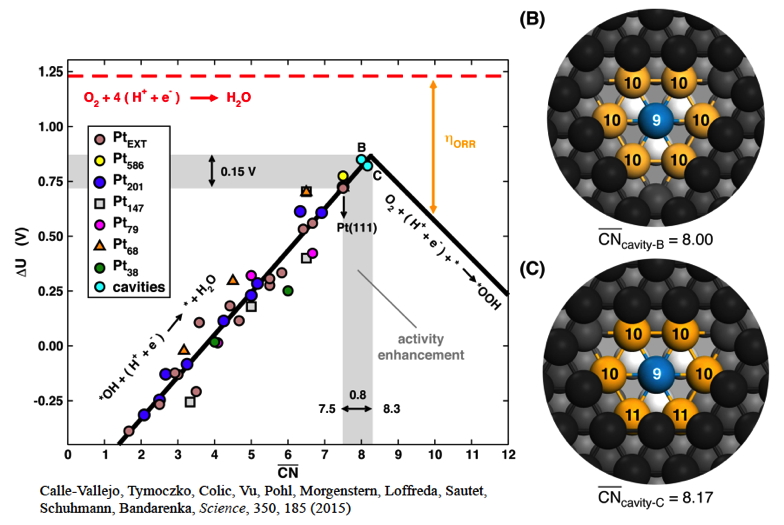
Catalyst optimization from coordination-activity volcano plot for oxygen reduction reaction on Pt. Cavities (B or C) are predicted to be more active. Selected Publications:
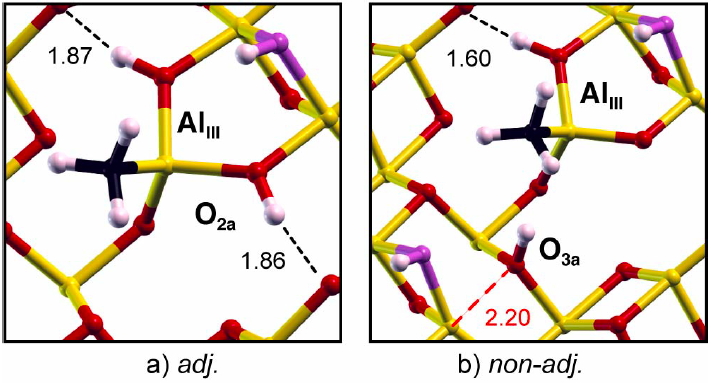
Calle-Vallejo, F.; Tymoczko, J.; Colic, V.; Vu, Q. H.; Pohl, M. D.; Morgenstern, K.; Loffreda, D.; Sautet, P.; Schuhmann, W.; Bandarenka, A. S. Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors. Science 2015, 350, 185-189. Read Calle-Vallejo, F.; Pohl, M. D.; Reinisch, D.; Loffreda, D.; Sautet, P.; Bandarenka, A. S. Why conclusions from platinum model surfaces do not necessarily lead to enhanced nanoparticle catalysts for the oxygen reduction reaction. Chem. Sci. 2017, DOI: 10.1039/C6SC04788B. Read Structure of catalytic surfaces in realistic conditions of pressure and temperature:In experimental conditions of heterogeneous catalysis, the structure of the catalytic interface is far from that in vacuum at 0 K. This is extremely important since the superficial structure and composition will completely determine the surface reactivity. We embrace this complexity and construct surface phase diagrams at relevant temperatures and pressures, based on DFT calculations, and incorporating in an explicit way the entropic contributions of gas phase species. An example of this is the surface of alumina Al2O3, a material heavily used in heterogeneous catalysis. We showed that the relevant surface is in fact hydroxylated. This allows for a detailed understanding of the chemistry of these surfaces and opens numerous applications (grafted complexes or growth of metallic particles on this oxide support).
Methane dissociation on an alumina 3-coordinated hydration defect Selected Publications:
M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, Use of DFT to achieve a rational understanding of acid-basic properties of gamma-alumina surfaces,Journal of Catalysis 2004, 226, 54-68. Read 
Surface palladium carbide formed in the reaction conditions of acetylene hydrogenation Selected Publications:
Transition metal clusters in catalysis:
Industrial catalysts are usually in the form of finely dispersed nanoparticles deposited on a large surface area oxide support. We study the structure and reactivity of transition metal clusters (Pt, Pd) deposited on oxides. Of a particular interest are the effects of the support, as well as pressures and resultant coverage with adsorbates, on the structure and electronic properties of these clusters. When submitted to a pressure of hydrogen, Pt13 deposited on gamma-alumina is covered by a large amount of hydrogen atoms (3 H atoms by surface Pt atoms, i.e. 3 times more than the Pt(111) surface) and its shape is modified from a biplanar to a cuboctahedral one. Again, the nature of the catalytic sites is very different from an isolated and bare cluster, if the system is thus treated in a realistic way.
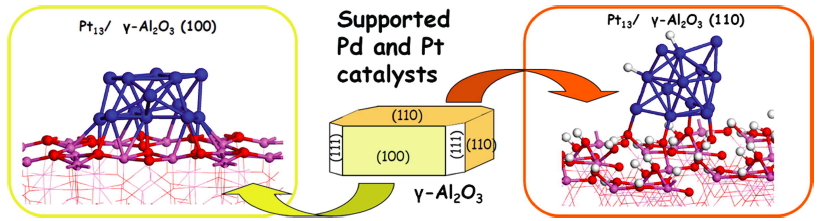
Structure of Pt13 on the non-hydroxylated (100) and on the hydroxylated (110) surfaces of gamma-alumina 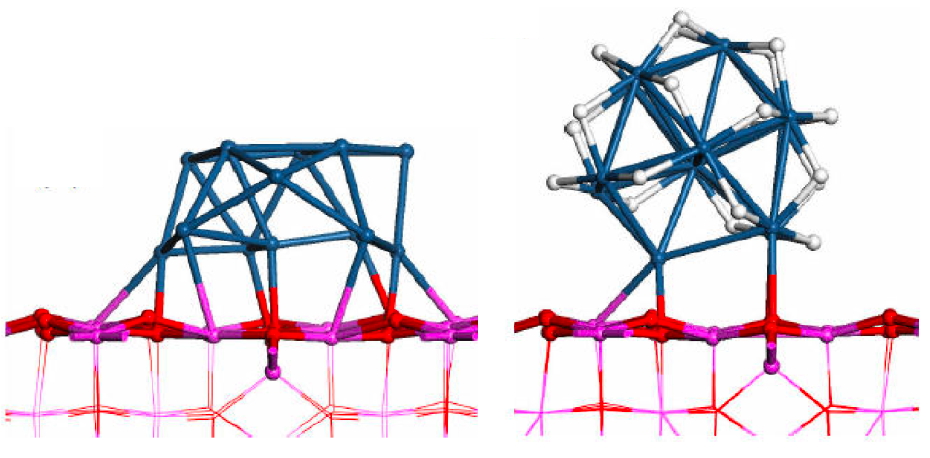
Shape of a Pt13 particle on the (100) surface of gamma-alumina in the absence (left) and presence (right) of hydrogen pressure, with 20 hydrogen atoms chemisorbed Selected Publications:
Semiconductors absorbing in the visible range for photocatalysis:
A key reaction of interest in the photocatalytic dissociation of water in hydrogen and oxygen, that yields clean fuel. The role of semiconductor is to absorb light, producing excitons, which dissociates into holes and electrons. The charge carriers then migrate to the surface or to a co-catalyst where the water oxidation reaction takes place. We use first-principle calculations to rationalize catalyst design. Besides band-gap, first-principles calculations allow us to determine the dielectric constant and the charge carrier mobility, which are key properties for a good dissociation of the exciton and migration of charges towards the catalytic sites. Several semiconductors have been studied (TiO2, Ta3N5, C3N4, ...) and doping strategies are considered.
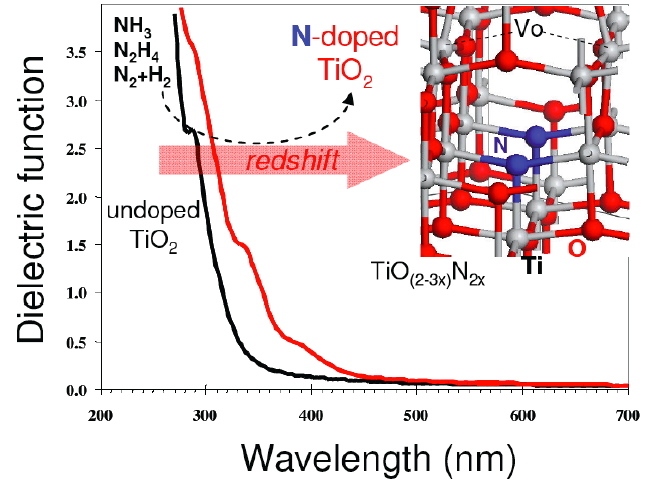
Adsorption spectrum calculated with DFT and the HSE06 functional for TiO2 (black) and for the structure doped with nitrogen (red). Selected publications:
E. Nurlaela, S. Ould-Chikh, M. Harb, S. del Gobbo, M. Aouine, E. Puzenat, P. Sautet, K. Domen, J.-M. Basset, and K. Takanabe, Critical role of the semiconductor-electrolyte interface in photocatalytic performance for water-splitting reactions using Ta3N5 particles, Chem. Mater. 2014, 26, 4812. Read T. Le Bahers, M. Rérat, P. Sautet, Semiconductors used in Photovoltaic and Photocatalytic devices: Assessing Fundamental Properties from DFT, J. Phys. Chem. C. 2014, 118, 5997-6008. Read Modeling electrocatalytic conditions from first principles:
We develop approaches for modeling electrochemical interfaces, where a surface is submitted to a potential and in contact with an electrolyte. Applications concern elementary reactions steps for electrocatalytic reactions (Formic acid electro-oxidation, reduction of CO2, electro-carboxylation of butadiene) and the influence of applied potential of molecular chemisorption modes.

Chemisorption of pyridine on Au(111) under oxidizing potential and distribution of counter-charge (in green) when modeling the electrolyte by the Linearized Poisson-Boltzmann equation Selected publications:
S. N. Steinmann, C. Michel, R. Schwiedernoch, J.-S. Filhol, P. Sautet, Modeling the HCOOH/CO2 Electrocatalytic Reaction: When Details Are Key, ChemPhysChem 2015, 16, 2307-2311. Read |