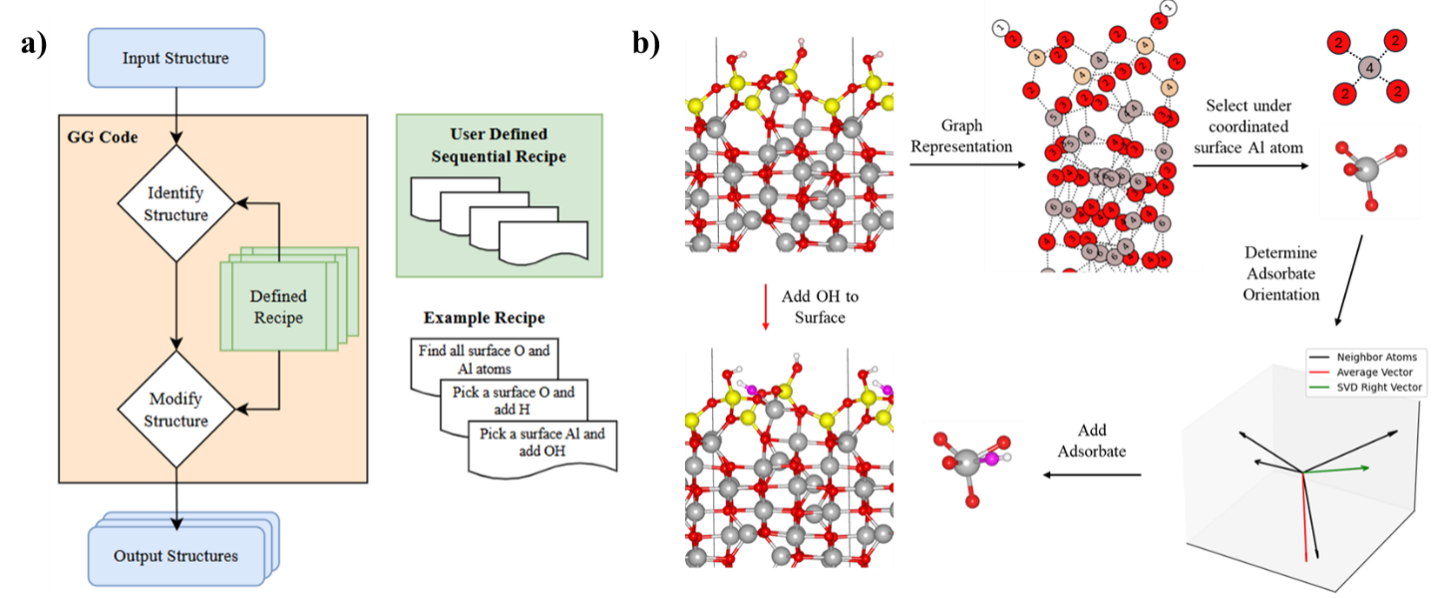
Structure prediction is a key challenge in computational modeling, particularly in surface catalysis, where atomic configurations determine active sites, reaction pathways, and activity. Global optimization (GO) methods such as simulated annealing, basin hopping, and genetic algorithms explore high-dimensional energy landscapes to identify low energy surface structures. However, integrating these methods with first-principles techniques like density functional theory (DFT) is computationally expensive. GO methods also struggle with defining precise control variables for structural exploration. Unbiased approaches (e.g., simulated annealing) require impractically high temperatures to overcome energy barriers, while biased methods (e.g., evolutionary algorithms) rely on chemical intuition, generating configurations through random perturbations or predefined transitions. These limitations hinder the systematic identification of catalytic structures under experimental conditions.
To address these challenges, we propose a graph-based framework that standardizes structural transitions (GitHub). Building on established graph representation tools, our approach extends to a wider range of catalytic systems. By integrating these tools with a grand canonical basin hopping GO algorithm, users can efficiently identify thermodynamically stable configurations under experimental conditions (e.g., temperature, pressure). The graph-based representation enhances search efficiency and provides a more accurate depiction of vibrational energies. This framework mitigates biases in conventional GO methods while remaining flexible for system-specific customization.