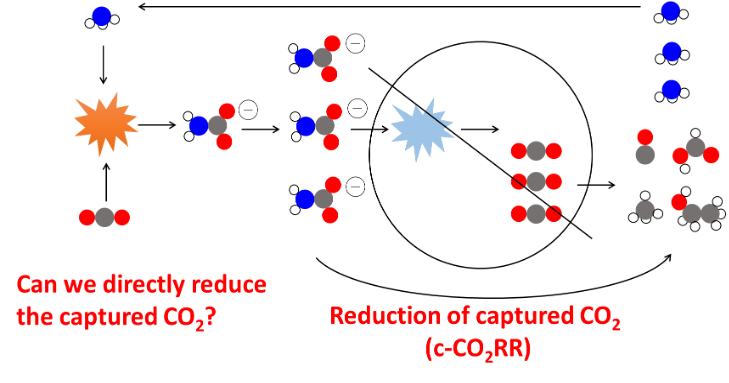
Since the industrial revolution the concentration of atmospheric CO₂ has risen drastically. Various methods to convert this CO₂ into higher value products have been proposed with one of the most promising being the CO₂ reduction reaction (CO₂RR). This process can be done both electrochemically and thermochemically, and the group has experience in both.
Traditionally, research focused on the CO₂RR assumes that the CO₂ has been concentrated. However, the CO₂ must be first captured and released, and this process is quite energy intensive. Thus, it has been proposed to directly reduce the captured CO₂ in the captured CO₂ reduction reaction (c-CO₂RR).
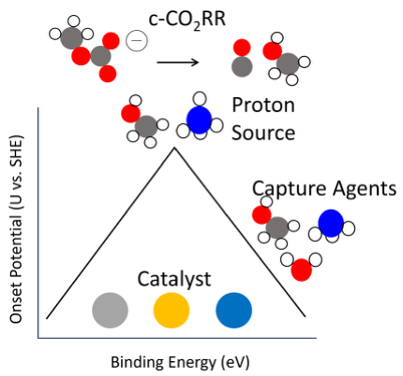
In the group we use ab-initio methods (density function theory) to computationally model the electrochemical c-CO₂RR reaction pathway. We have experience in both thermodynamics and kinetic simulations to model the reactivity of the c-CO₂RR, CO₂RR and the competitive hydrogen evolution reaction (HER). To our knowledge, our group published the first complete cycle of the c-CO₂RR to CO. 2 We determine that the c-CO₂RR requires 3 protonations rather than the 2 needed in the CO₂RR, and thus, we found that the identity of the proton source greatly affects the activity c-CO₂RR. We find that with an NH₄⁺ cation as a proton source, the Ag(111) is more active for c-CO₂RR than the CO₂RR. However, we saw the HER also is highly proton source dependent and therefore, we determined that it was imperative to look for catalysts that significantly hinder the HER. Interestingly, we find that the capture agent has little effect on the overall reactivity. Thus, we find that one must choose a strong proton source and a catalyst that severely hinders the HER for the catalyst to be selective for c-CO₂RR.