Ab Initio Molecular Cluster Calculation
At UCLA, we have developed a method of simulating reactions on
compound semiconductor surfaces using molecular cluster calculations with density
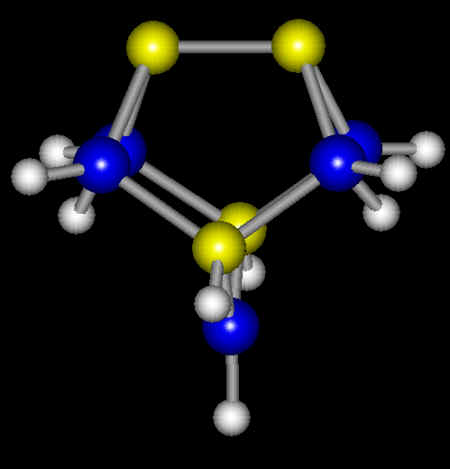
functional theory. Shown below is a cluster
model of the gallium arsenide surface. This
cluster represents all the reaction sites on the surface, i.e., an arsenic dimer and two
second-layer gallium atoms. On the optimized
cluster, each arsenic dangling bond is filled with a pair of electrons, while each gallium
dangling bond is empty. This is in excellent
agreement with the experimental observations. The
most exciting result from this work is that we can predict the vibrational frequencies of
the optimized clusters and compare these results with our infrared data. This unique capability allows us to definitely
assign the observed vibrational bands to specific adsorption sites. Furthermore, I have extended this method to study
the reaction mechanisms of organometallic precursors on the semiconductor surfaces.
More on molecular clusters:
- Decomposition mechanism of arsine on GaAs (001)-(4x2)
- InP (001)-(2x1) and its spin density distribution
- Hydrogen adsorption on InP (001)-(2x1)
- Hydrogen adsorption on InP (001)-(2x4)